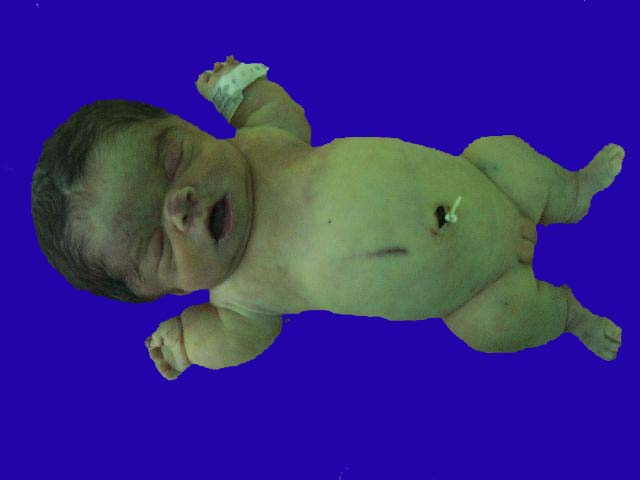
El término mosaicismo se utiliza para describir la presencia de más de un tipo de célula en un individuo. Por ejemplo, una persona puede tener algunas de las células de su cuerpo con 46 cromosomas, mientras que otras células de su cuerpo pueden tener 47 cromosomas. Un ejemplo de mosaicismo en el síndrome de Down con alteración cromosómica en mosaico.
Aproximadamente el 95 por ciento de las personas que padecen síndrome de Down tienen trisomía 21, es decir, un cromosoma 21 adicional en cada célula de su cuerpo. Entre un 3 y un 4 por ciento de los individuos con síndrome de Down padecen síndrome de Down por translocación. Esto significa que el cromosoma 21 adicional, o parte de él, está adosado a otro cromosoma. El 1 y 2 por ciento restante de individuos que padecen síndrome de Down tienen una alteración cromosómica en mosaico. Por consiguiente, se deduce que tienen por lo menos dos tipos de células; algunas con el número normal de cromosomas (un total de 46) y otras con un cromosoma 21 adicional (un total de 47). En raras oportunidades, una persona puede tener más de dos tipos de líneas celulares.
El mosaicismo generalmente se describe por medio de un porcentaje. Por ejemplo, cuando nace un bebé con síndrome de Down, el médico le toma una muestra de sangre para realizar un estudio cromosómico. Por lo general, se analizan 20 células diferentes. Si cinco de esas 20 células son normales (46 cromosomas) y las 15 restantes tienen un cromosoma 21 adicional (47 cromosomas), se determina que el bebé tiene síndrome de Down con alteración cromosómica en mosaico. Dado que el porcentaje de células con un cromosoma adicional es 15 sobre un total de 20, se establecerá que el bebé tiene un nivel de mosaicismo del 75 por ciento. Los porcentajes pueden variar en las distintas partes del cuerpo. El porcentaje de células trisómicas en el músculo puede ser distinto del porcentaje registrado en el cerebro o el porcentaje registrado en la sangre o la piel.
Generalmente, un bebé recibe la mitad de sus cromosomas de cada progenitor, de manera que cada óvulo y espermatozoide tiene 23 cromosomas. Luego de la fertilización, ocurre un proceso rápido de división celular por el cual una célula duplica sus cromosomas (realiza una copia de ellos) y luego se divide por la mitad. En otras palabras, la célula pasa de tener 46 cromosomas a tener 92 cromosomas y luego se divide por la mitad, resultando en dos células nuevas de 46 cromosomas cada una. Este tipo de división celular se denomina mitosis.
En algún momento posterior a la fertilización, se puede producir un error en la mitosis, es decir, una célula tal vez no se divida exactamente en dos mitades. Como consecuencia de ello, algunas células tendrán el número normal o 46 cromosomas mientras que otras tendrán un cromosoma 21 adicional o 47 cromosomas.
Aproximadamente el 95 por ciento de las personas que padecen síndrome de Down tienen trisomía 21, es decir, un cromosoma 21 adicional en cada célula de su cuerpo. Entre un 3 y un 4 por ciento de los individuos con síndrome de Down padecen síndrome de Down por translocación. Esto significa que el cromosoma 21 adicional, o parte de él, está adosado a otro cromosoma. El 1 y 2 por ciento restante de individuos que padecen síndrome de Down tienen una alteración cromosómica en mosaico. Por consiguiente, se deduce que tienen por lo menos dos tipos de células; algunas con el número normal de cromosomas (un total de 46) y otras con un cromosoma 21 adicional (un total de 47). En raras oportunidades, una persona puede tener más de dos tipos de líneas celulares.
El mosaicismo generalmente se describe por medio de un porcentaje. Por ejemplo, cuando nace un bebé con síndrome de Down, el médico le toma una muestra de sangre para realizar un estudio cromosómico. Por lo general, se analizan 20 células diferentes. Si cinco de esas 20 células son normales (46 cromosomas) y las 15 restantes tienen un cromosoma 21 adicional (47 cromosomas), se determina que el bebé tiene síndrome de Down con alteración cromosómica en mosaico. Dado que el porcentaje de células con un cromosoma adicional es 15 sobre un total de 20, se establecerá que el bebé tiene un nivel de mosaicismo del 75 por ciento. Los porcentajes pueden variar en las distintas partes del cuerpo. El porcentaje de células trisómicas en el músculo puede ser distinto del porcentaje registrado en el cerebro o el porcentaje registrado en la sangre o la piel.
Generalmente, un bebé recibe la mitad de sus cromosomas de cada progenitor, de manera que cada óvulo y espermatozoide tiene 23 cromosomas. Luego de la fertilización, ocurre un proceso rápido de división celular por el cual una célula duplica sus cromosomas (realiza una copia de ellos) y luego se divide por la mitad. En otras palabras, la célula pasa de tener 46 cromosomas a tener 92 cromosomas y luego se divide por la mitad, resultando en dos células nuevas de 46 cromosomas cada una. Este tipo de división celular se denomina mitosis.
En algún momento posterior a la fertilización, se puede producir un error en la mitosis, es decir, una célula tal vez no se divida exactamente en dos mitades. Como consecuencia de ello, algunas células tendrán el número normal o 46 cromosomas mientras que otras tendrán un cromosoma 21 adicional o 47 cromosomas.