

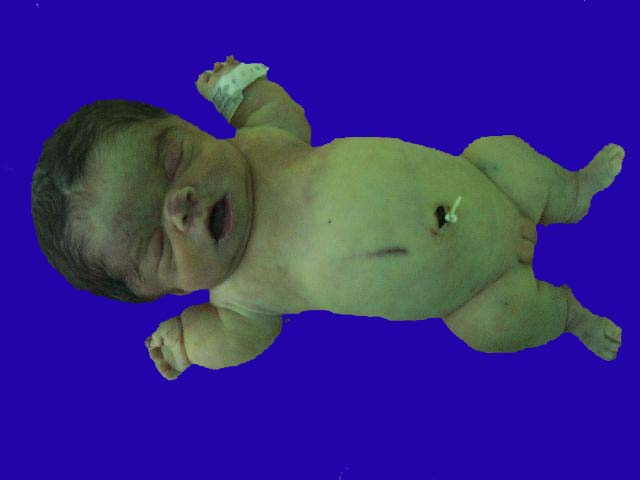
¿QUE ES LA ACONDROPLASIA?
La acondroplasia es una enfermedad genética que se presenta en 1 de cada 25.000 niños nacidos vivos. Se trata de un trastorno del crecimiento óseo; de hecho, el nombre de la enfermedad proviene de 3 vocablos griegos (a = sin; chondro = cartílago; plasia = crecimiento o desarrollo), es decir, significa sin crecimiento normal del cartílago. Es el tipo más frecuente de enanismo que existe, caracterizado por un acortamiento de los huesos largos y mantenimiento de la longitud de la columna vertebral, lo que da un aspecto un tanto desarmónico: macrocefalia, piernas y brazos cortos y un tamaño normal del tronco, entre otras desirregularidades fenotípicas.
etiologia
La causa de esta enfermedad es una mutación en el gen que codifica para el receptor 3 del factor de crecimiento fibroblástico (FGFR3), localizado en el cromosoma 4. Existen dos mutaciones posibles que afectan a este gen: G1138A y G1138C. Ambas son mutaciones puntuales, donde dos pares de bases complementarias del DNA se intercambian:
Mutación G1138A: en el nucleótico número 1138, la guanina es sustituida por adenina. En el 98% de los casos de acondroplasia, se sufre esta mutación.
Mutación G1138C: tiene lugar el cambio de guanina por citosina, también en el nucleótido 1138. La frecuencia de esta alteración es mucho menor, apenas en el 2% de los casos.
En ambas situaciones, la repercusión en la cadena aminoacídica de la proteína FGFR3, es la misma: el cambio del aminoácido arginina por una glicina.
Dicha mutación puede darse de dos formas distintas: por herencia autosómica dominante, cuando hay antecedentes familiares de enfermedad (alrededor del 10% de los casos) y por una mutación de novo, sin padres enfermos (es la causa más frecuente, hasta en el 90% de los enfermos).
Herencia genética
La herencia de la enfermedad es autosómica dominante, lo que significa que, para padecer la enfermedad, basta con que se herede el gen mutado de, al menos, uno de los progenitores. Las posibilidades genotípicas y su correspondencia fenotípica, son las siguientes:
Homocigoto enfermo (G1138A/G1138A): es la forma más grave de la enfermedad y suele ser letal durante el período neonatal. Para que tenga lugar este hecho, es necesario que ambos progenitores sean acondroplásicos (heterocigotos, pues los homocigotos no sobreviven) y se da en el 25% de las parejas enfermas.
Heterocigoto (G1138A/alelo normal): a este genotipo se puede llegar desde dos supuestos posibles. Si ambos padres están enfermos, la posibilidad de que el hijo sea heterocigoto para la enfermedad es de un 50%; pero también hay un 50% de posibilidades de heredar la enfermedad cuando sólo uno de los progenitores tiene acondroplasia.
Homocigoto sano (alelo normal/alelo normal): desde el punto de vista mendeliano, tres son las posibilidades: 1. Un 25% de probabilidad cuando los dos padres son acondroplásicos. 2. Si sólo uno de los progenitores está enfermo, la probabilidad sube al 50%. 3. Ambos padres sanos.
Mutación G1138A: en el nucleótico número 1138, la guanina es sustituida por adenina. En el 98% de los casos de acondroplasia, se sufre esta mutación.
Mutación G1138C: tiene lugar el cambio de guanina por citosina, también en el nucleótido 1138. La frecuencia de esta alteración es mucho menor, apenas en el 2% de los casos.
En ambas situaciones, la repercusión en la cadena aminoacídica de la proteína FGFR3, es la misma: el cambio del aminoácido arginina por una glicina.
Dicha mutación puede darse de dos formas distintas: por herencia autosómica dominante, cuando hay antecedentes familiares de enfermedad (alrededor del 10% de los casos) y por una mutación de novo, sin padres enfermos (es la causa más frecuente, hasta en el 90% de los enfermos).
Herencia genética
La herencia de la enfermedad es autosómica dominante, lo que significa que, para padecer la enfermedad, basta con que se herede el gen mutado de, al menos, uno de los progenitores. Las posibilidades genotípicas y su correspondencia fenotípica, son las siguientes:
Homocigoto enfermo (G1138A/G1138A): es la forma más grave de la enfermedad y suele ser letal durante el período neonatal. Para que tenga lugar este hecho, es necesario que ambos progenitores sean acondroplásicos (heterocigotos, pues los homocigotos no sobreviven) y se da en el 25% de las parejas enfermas.
Heterocigoto (G1138A/alelo normal): a este genotipo se puede llegar desde dos supuestos posibles. Si ambos padres están enfermos, la posibilidad de que el hijo sea heterocigoto para la enfermedad es de un 50%; pero también hay un 50% de posibilidades de heredar la enfermedad cuando sólo uno de los progenitores tiene acondroplasia.
Homocigoto sano (alelo normal/alelo normal): desde el punto de vista mendeliano, tres son las posibilidades: 1. Un 25% de probabilidad cuando los dos padres son acondroplásicos. 2. Si sólo uno de los progenitores está enfermo, la probabilidad sube al 50%. 3. Ambos padres sanos.
No hay comentarios:
Publicar un comentario